Pipeline
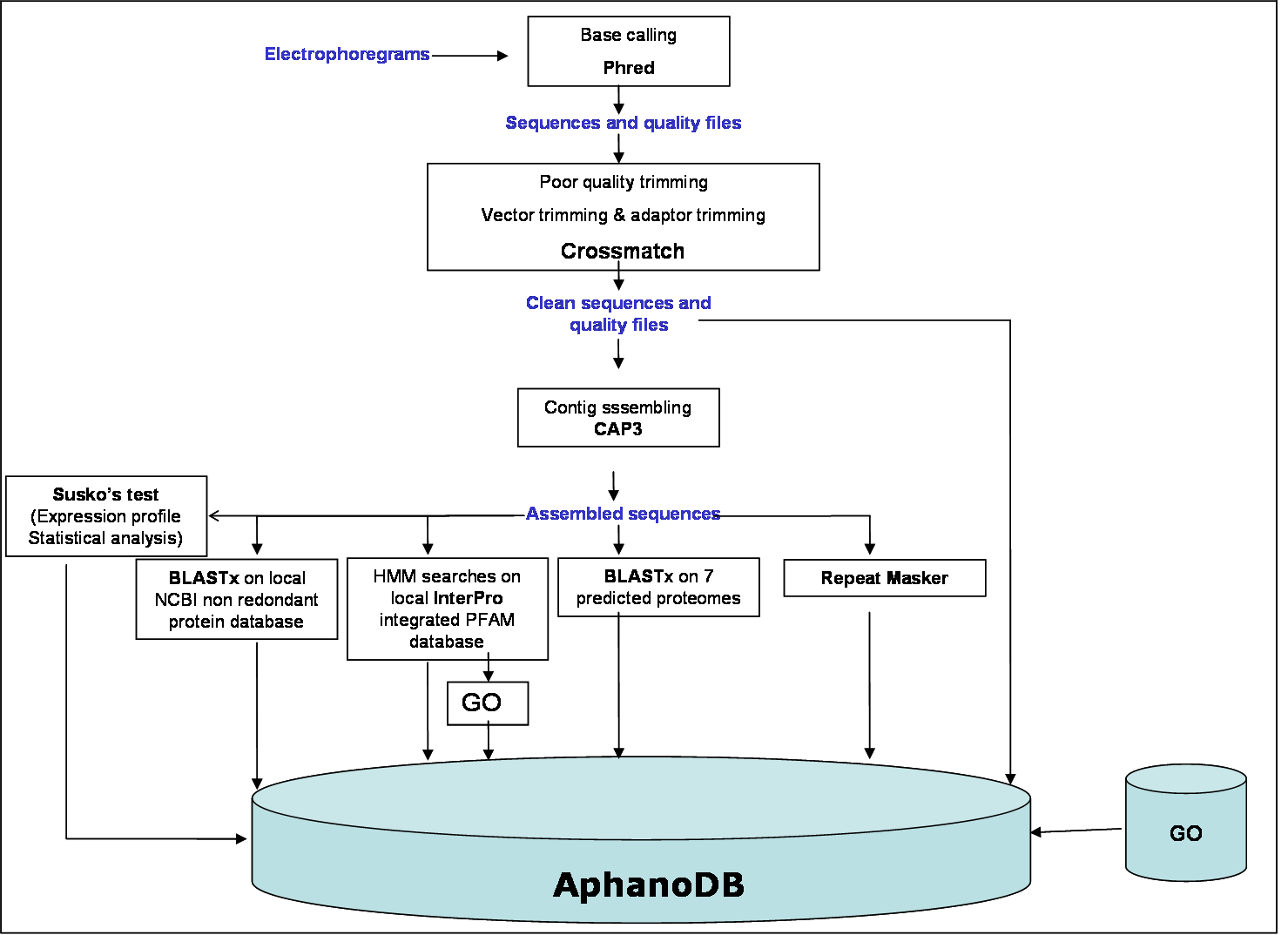
EST processing and assembly :
All reads were obtained using the Phred program. Multifasta sequence and quality files were generated and cleaned by vector and adaptor trimming using crossmatch. Only reads with 80% of 20 Phred value and with a length over 100 bp were selected and 18684 successful sequences were obtained. High quality consensus sequences were assembled using CAP3 program. To reach a good assembling minimum overlap length cutoff was set to 100 bp, minimum identity percent to 97% and maximum gap length to 30 bp (-o 100 -p 97 -f 30).
ESTs of strain ATCC 201684 from the two libraries (interaction and saprophytic) were assembled together to obtain finally 7977 unigenes composed of 2843 contigs and 5134 singletons. Consensus sequences were renamed with a unique identifier with the prefix 'Ae' for the species, 3 digits for the number of EST included in the contig, 2 digits for the strain ('AL' for strain ATCC 201684) and 5 digits as unique number.
Sequence comparison and assignment into functional categories :
First, assignment of putative function was performed by running the BLASTx algorithm against a local non-redundant protein database (NR, NCBI, release 2.2.14). Filter of low complexity was used.
Secondly, all sequences were compared with BLASTx algorithm to seven predicted proteome banks (Arabidopsis thaliana, Toxoplasma gondii, Plasmodium falciparum, Thalassiosira pseudonana, Phytophthora sojae, Phytophthora ramorum, and fusarium solani). For each bank the blastx results were stored in AphanoDB, and each proteic sequences of each bank were also stored.
Then, domain searches using InterPro database (release 4.2) were made. HMM searches against Pfam protein database (16.0) were performed locally.
To classify the sequence according to the Gene Ontology, Pfam domain with InterPro ID were linked to GO molecular function, biological process and cellular component terms using interpro2go index file (ebiftp://ftp.ebi.ac.uk/pub/databases/interpro).